Introduction to MAGEMin_C
Table of Contents
Requirements
This tutorial assumes you have
Visual Studio CodeandJuliainstalled.Detailed documentation is provided here
Installation & Setup
Create a dedicated local folder for MAGEMin_C to live in its own environment. This avoids potential conflicts with other packages such as MAGEMinApp.
1. Create the working directory
cd /where_you_want_your_folder
mkdir MAGEMin_tutorial
cd MAGEMin_tutorial
julia
2. Install packages
Open the Julia package manager by pressing ], then activate the local environment and add the required packages:
]
activate .
add MAGEMin_C, Plots
This installs both MAGEMin_C and Plots, the latter being the visualization package used throughout this tutorial. More advanced alternatives such as Makie or PlotlyJS exist but are beyond the scope of this introduction.
Note
activate . creates a local Julia environment in MAGEMin_tutorial/ where MAGEMin_C and Plots are installed. If you close the terminal, you will need to re-activate it (] activate .) to reload the local environment.
First minimization script
1. Create a new Julia script
Simply open a new terminal in the MAGEMin_tutorial folder and type:
code first_minimization.jlThis creates a Julia script (.jl) and opens the file in VS Code.
2. Load MAGEMin_C and Plots packages
In the empty file, add the following line at the top of the file:
using MAGEMin_C, Plots3. Initialize MAGEMin
data = Initialize_MAGEMin("mp", verbose=false);This command initializes MAGEMin using the "mp" database (or a-x set of models) which stands for "metapelite" (White et al., 2014).
Available thermodynamic a-x sets:
| Acronym | Dataset | Reference |
|---|---|---|
mtl | Mantle | Holland et al., 2013 |
mp | Metapelite | White et al., 2014 |
mb | Metabasite | Green et al., 2016 |
ig | Igneous | Green et al., 2025 (updated from Holland et al., 2018) |
igad | Igneous alkaline dry | Weller et al., 2024 |
um | Ultramafic | Evans & Frost, 2021 |
sb11 | Stixrude & Lithgow-Bertelloni | 2011 |
sb21 | Stixrude & Lithgow-Bertelloni | 2021 |
sb24 | Stixrude & Lithgow-Bertelloni | 2024 |
ume | Ultramafic extended | Green et al., 2016 + Evans & Frost, 2021 |
mpe | Extended metapelite | White et al., 2014 + Green et al., 2016 + Franzolin et al., 2011 + Diener et al., 2007 |
mbe | Extended metabasite | Green et al., 2016 + Diener et al., 2007 + Rebay et al., 2022 |
4. Provide system information
Provide a composition and a system unit:
X = [0.5922, 0.1813, 0.006, 0.0223, 0.0633, 0.0365, 0.0127, 0.0084, 0.0016, 0.0007, 0.075]
Xoxides = ["SiO2", "Al2O3", "CaO", "MgO", "FeO", "K2O", "Na2O", "TiO2", "O", "MnO", "H2O"]
sys_unit = "wt"Here, we use a water oversaturated average pelite composition. Note that sys_unit can be "wt" or "mol".
Define pressure and temperature:
P = 10.0
T = 700.05. Call single point minimization routine
out = single_point_minimization(P, T, data, X=X, Xoxides=Xoxides, sys_in=sys_unit, name_solvus = true)Here, the argument name_solvus automatically tries to correctly name several instances of the same solution model based on their composition. For instance, feldspar can be either plagioclase pl or alkali-feldspar afs.
At this stage, your first MAGEMin script should look like:

6. Perform the stable phase equilibrium calculation
To execute the script, either copy and paste the content of your script in the Julia terminal or type:
include("first_minimization.jl")This should result in:
julia> include("first_minimization.jl")
Pressure : 10.0 [kbar]
Temperature : 700.0 [Celsius]
Stable phase | Fraction (mol fraction)
g 0.07753
mu 0.169
liq 0.24053
bi 0.09972
ilm 0.01863
q 0.24848
ky 0.04752
ru 0.00037
H2O 0.09822
Stable phase | Fraction (wt fraction)
g 0.09343
mu 0.19963
liq 0.20529
bi 0.10707
ilm 0.02116
q 0.27091
ky 0.06987
ru 0.00054
H2O 0.03211
Stable phase | Fraction (vol fraction)
g 0.0595
mu 0.18
liq 0.25537
bi 0.09142
ilm 0.01096
q 0.26397
ky 0.04914
ru 0.00033
H2O 0.0893
Gibbs free energy : -853.149024 (27 iterations; 16.83 ms)
Oxygen fugacity : -13.512530621551143
Delta QFM : 2.495598922485575The quick output shows you a summary of the results of the calculation, providing you with the stable phases and their mol, wt and vol fractions.
7. Access minimization results
The full output of the calculation is saved in the out structure. The different fields can be accessed in the terminal by typing out. and pressing "tab":
out.which unfolds to:
out.G_system Gamma MAGEMin_ver M_sys PP_syms PP_vec P_kbar SS_syms SS_vec T_C V Vp Vp_S Vs Vs_S X
aAl2O3 aFeO aH2O aMgO aSiO2 aTiO2 alpha buffer buffer_n bulk bulkMod bulkModulus_M bulkModulus_S bulk_F bulk_F_wt bulk_M
bulk_M_wt bulk_S bulk_S_wt bulk_res_norm bulk_wt dQFM database dataset elements enthalpy entropy eta_M fO2 frac_F frac_F_vol frac_F_wt
frac_M frac_M_vol frac_M_wt frac_S frac_S_vol frac_S_wt iter mSS_vec n_PP n_SS n_mSS oxides ph ph_frac ph_frac_1at ph_frac_vol
ph_frac_wt ph_id ph_id_db ph_type rho rho_F rho_M rho_S s_cp shearMod shearModulus_S sol_name status time_msA full description of every variable and array stored in the out structure is provided here. This includes detailed information on how to access the composition of phases in different units (vol, mol, wt, apfu), fraction but also other properties such as heat capacity, density, seismic velocity and melt viscosity (among others).
For instance, stable phases acronyms can be accessed as:
out.ph9-element Vector{String}:
"g"
"mu"
"liq"
"bi"
"ilm"
"q"
"ky"
"ru"
"H2O"...and the mole fraction of stable melt can be accessed as:
out.frac_M0.2405316872497568while the volume fraction as:
out.frac_M_vol0.2553733579195757where "M", stands for melt and can be replaced by "S" solid, and "F" for fluid.
8. Retrieve garnet composition in wt
To extract the composition of garnet, one needs to access the properties of the "g" phase which are stored in out.SS_vec.
- Note that
SS_vecis a sub structure ofoutthat stores all the information about the stable solution phases. Instead, the information about the pure phases are accessed inPP_vecsub-structure.
Because SS_vec is an array of structure of size n solution phase, there are several ways to retrieve the index of garnet. For instance, one could look for the index of garnet in out.ph
id_g = findfirst(out.ph .== "g")1then access the information of garnet using id_g as:
out.SS_vec[id_g].Comp Comp_apfu Comp_wt G V Vp
Vs alpha bulkMod compVariables compVariablesNames cp
deltaG emChemPot emComp emComp_apfu emComp_wt emFrac
emFrac_wt emNames enthalpy entropy f rho
shearMod siteFractions siteFractionsNamesand its composition in wt as:
out.SS_vec[id_g].Comp_wt11-element Vector{Float64}:
0.38117580289882985
0.2026163965532957
0.04357834582077847
0.06089592806492956
0.3024411056038425
0.0
0.0
0.0
0.002041744392071351
0.007250676666252689
0.0Warning
The ordering of the output oxides may differ from the input ones. This is because MAGEMin internally reorganizes them. To check the ordering of the oxides in the out structure simply type:
out.oxides11-element Vector{String}:
"SiO2"
"Al2O3"
"CaO"
"MgO"
"FeO"
"K2O"
"Na2O"
"TiO2"
"O"
"MnO"
"H2O"In a similar manner, the composition in atom per formula unit can be accessed as:
out.SS_vec[id_g].Comp_apfuwhich gives:
11-element Vector{Float64}:
2.9999999999999996
1.8793194615095314
0.36744182626293087
0.7145112355900585
1.9903978756455607
0.0
0.0
0.0
12.060340269245234
0.048329600991918464
0.09. Plot garnet composition
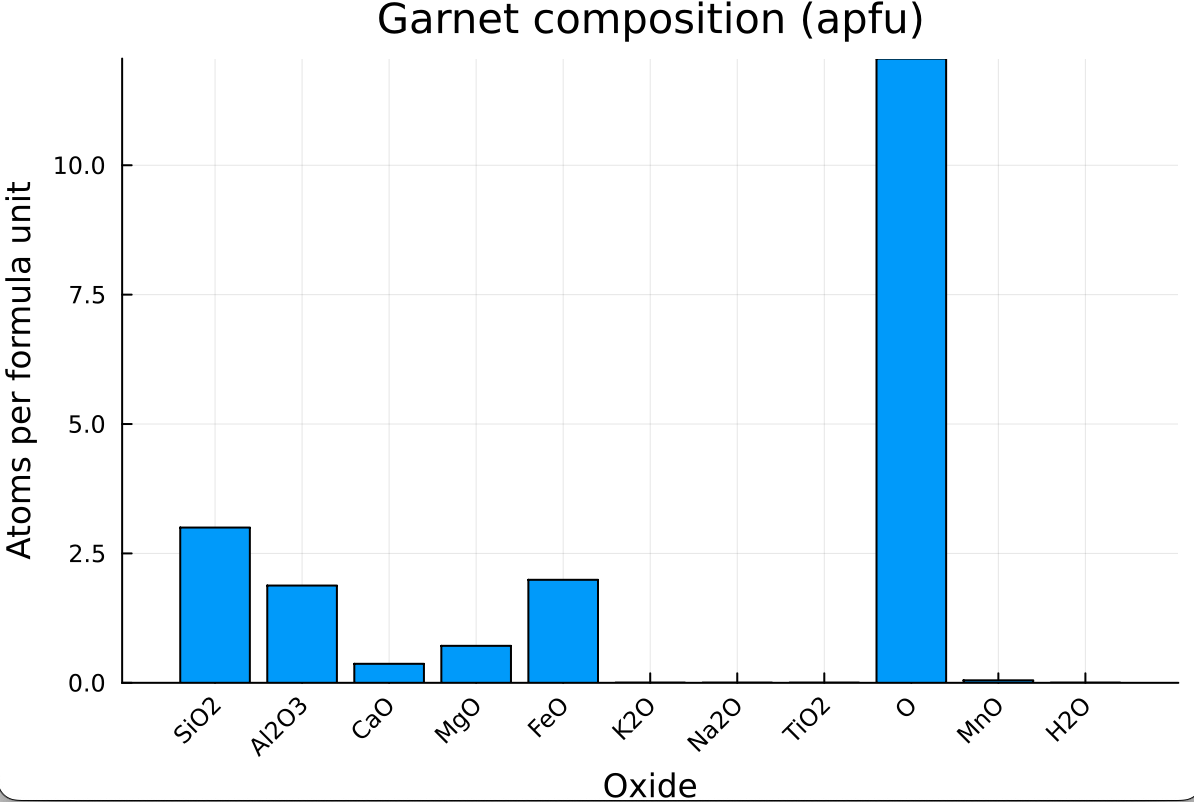
Using Plots.jl, we can visualize the garnet composition (in apfu) as a bar chart where each bar is labeled by its oxide:
bar(out.oxides, out.SS_vec[id_g].Comp_apfu,
xlabel = "Oxide",
ylabel = "Atoms per formula unit",
title = "Garnet composition (apfu)",
legend = false,
xrotation = 45)which should give:
10. Plot melt and solid bulk composition
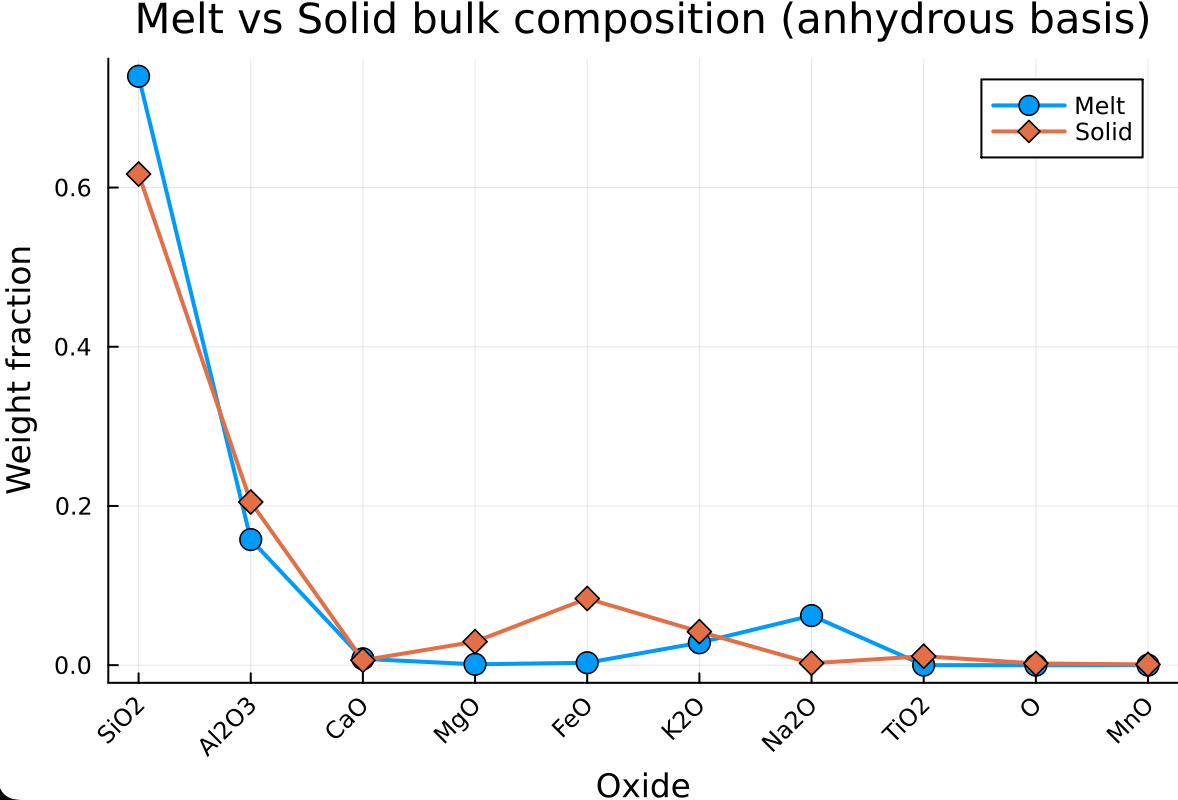
We can compare the bulk composition of the melt and solid sub-systems on an anhydrous basis using a scatter + line diagram:
first renormalize the composition of the solid and the melt in an anhydrous basis:
dry_oxides = findall(out.oxides .!= "H2O")
bulk_M_wt_dry = out.bulk_M_wt[dry_oxides] ./ sum(out.bulk_M_wt[dry_oxides])
bulk_S_wt_dry = out.bulk_S_wt[dry_oxides] ./ sum(out.bulk_S_wt[dry_oxides])plot(out.oxides[dry_oxides], bulk_M_wt_dry,
label = "Melt",
markershape = :circle,
markersize = 6,
linewidth = 2,
xlabel = "Oxide",
ylabel = "Weight fraction",
title = "Melt vs Solid bulk composition (anhydrous basis)",
xrotation = 45)
plot!(out.oxides[dry_oxides], bulk_S_wt_dry,
label = "Solid",
markershape = :diamond,
markersize = 6,
linewidth = 2)which should give:
Exercises
E.1. Calculate residual composition
Assuming that any volume of melt > 0.07 vol_fraction is extracted, as well as any free water is extracted, calculate the remaining residual bulk-rock composition in
wtfraction.Mind that
Densities of solid, melt and fluid can be accessed as
out.rho_S,out.rho_Mandout.rho_FSolid, melt and fluid
wtcompositions can be accessed asout.bulk_S_wt,out.bulk_M_wtandout.bulk_F_wt
Using the plot from point #10 (Plot melt and solid bulk composition), add the composition of the residual to the plot