MAGEMin_C.jl: Tutorials
This page provides a set of detailed tutorials showing how to use MAGEMin_C.jl to perform phase equilibrium calculations.
Info
Note
- The tutorials are not optimized for performances, but are provided in hope they can be useful to present
MAGEMin_Cfunctionality.
Iterative phase equilibrium calculation
first add MAGEMin_C and Plots
julia> ] add MAGEMin_C
julia> ] add Plotsand use MAGEMin_C and Plots as:
using MAGEMin_C, PlotsFirst let's first initialize MAGEMin with the metapelite database (White et al ., 2014)
data = Initialize_MAGEMin("mp", verbose=false);Then define the bulk-rock composition (wt fraction) the related oxide list and the system unit
X = [0.5922, 0.1813, 0.006, 0.0223, 0.0633, 0.0365, 0.0127, 0.0084, 0.0016, 0.0007, 0.075]
Xoxides = ["SiO2", "Al2O3", "CaO", "MgO", "FeO", "K2O", "Na2O", "TiO2", "O", "MnO", "H2O"]
sys_unit = "wt"Note
Here we use the water-oversaturated FWorld Average composition for metapelite
Define test pressure and temperature condition for test point:
P = 10.0
T = 700.0and perform the test calculation:
out = single_point_minimization(P, T, data, X=X, Xoxides=Xoxides, sys_in=sys_unit, name_solvus = true)Note
The option name_solvus = true is important here, as it allows for to properly name minerals based on their composition, e.g., fsp -> pl or afs
which should gives:
Pressure : 10.0 [kbar]
Temperature : 700.0 [Celsius]
Stable phase | Fraction (mol fraction)
g 0.07753
mu 0.169
liq 0.24053
bi 0.09972
ilm 0.01863
q 0.24848
ky 0.04752
ru 0.00037
H2O 0.09821
Stable phase | Fraction (wt fraction)
g 0.09343
mu 0.19964
liq 0.20529
bi 0.10706
ilm 0.02116
q 0.27091
ky 0.06987
ru 0.00054
H2O 0.03211
Stable phase | Fraction (vol fraction)
g 0.0595
mu 0.18
liq 0.25537
bi 0.09142
ilm 0.01096
q 0.26397
ky 0.04914
ru 0.00033
H2O 0.0893
Gibbs free energy : -853.149024 (27 iterations; 12.19 ms)
Oxygen fugacity : -13.51247569580698
Delta QFM : 2.4956538482297375Instead, of using a single pressure and temperature conditions let's now keep the pressure fixed and vary the temperature
n = 50
P = 10.0
T = collect(range(400.0,800.0,n))Here range(min,max,n) will create a range of value between min and max with n steps. collect() turns the range into an array that can be indexed.
Create a loop from 1 to n in order to compute a stable phase equilibrium for all the entries of the T array. This can be done as follow:
for i=1:n
T_calc = T[i] # retrieves the temperature from the temperature array we just defined
out = single_point_minimization(P, T_calc, data, X=X, Xoxides=Xoxides, sys_in=sys_unit)
endAlthough this performs the set of 50 phase equilibrium calculations as intended, the script is not yet very useful as the results of the calculation are not stored.
To store the results of the stable phase equilibrium calculations you can declare an array of MAGEMin_C output structure such as:
out = Vector{out_struct}(undef,n)
for i=1:n
T_calc = T[i] # retrieves the temperature from the temperature array we just defined
out[i] = single_point_minimization(P, T_calc, data, X=X, Xoxides=Xoxides, sys_in=sys_unit, name_solvus = true)
endNote
The first line of previous snippet create a Vector of MAGEMin_C structures, of size nand with undefined content. In this case we are interested in 1D array, but in a similar manner you could create a Matrix e.g., out = Matrix{out_struct}(undef,n,n)
Once the calculation are peformed you can access all the informations:
out[2]. #then hit double `tabulation` key twiceThe latter will display all the entries of point 2. If you want to retrieve the melt volume fraction you can do so by accessing out[2].frac_M_vol. In the case of point 2, we get:
julia> out[2].frac_M_vol
0.0Now the question is how to gather in a efficient juliaeskmanner the volume fractions for all computed points? One relatively quick and efficient way is as follow:
frac_M_vol = [out[i].frac_M_vol for i=1:n]which should yield in the terminal:
julia> frac_M_vol = [out[i].frac_M_vol for i=1:n]
50-element Vector{Float64}:
0.0
0.0
0.0
0.0
0.0
⋮
0.66483750887145
0.6823915840521158
0.7005692654225463
0.7193913179922915
0.7388665024379735To retrieve the vol fraction of a solid phase, the process is slightly more complex as we have to check in the phase is present in the mineral assemblage first. Let's first allocate an array for the volume fraction of quartz (q):
q_vol = zeros(Float64,n)Let us now loop through the output structure to look for quartz in the stable mineral assemblage, and in the event quartz is stable, retrieve its volume fraction:
for i=1:n
if "q" in out[i].ph #here we check if out[i].ph contains "q"
id_q = findfirst(out[i].ph .== "q")
q_vol[i] = out[i].ph_frac_vol[id_q]
end
endNote
We first here check if "q" is in the phase assemblage. Then the command id_q = findfirst(out[i].ph .== "q")find the position of "quartz" in the array to be able to retrieve to right value.
Using plots.jl, plot the volume fraction of melt and quartz as function of the temperature. Note that you can save the figure by using:
plot( T, q_vol .* 100,
label = "qtz",
xlabel = "T°C",
ylabel = "vol%")Note
You can save the figure using;
plot!(size=(400,400))
savefig("figure.png")The first line update the resolution of the plot according to your needs, while the second line effectively save the figure. Note that you can use different output format (png,jpg,pdf)
One way to make the model more realistic is by dynamically adjusting the composition at every step by removing excess water from it. This can be be done by modifying your calculation as follow:
if "H2O" in out[i].ph
id_h2o = findfirst(out[i].ph .== "H2O")
h2o_wt = out[i].ph_frac_wt[id_h2o]
h2o_comp_wt = out[i].PP_vec[id_h2o - out[i].n_SS].Comp_wt
X = X .- (h2o_wt .* h2o_comp_wt)
endHere, we first look for the id of "H2O" pure phase. Then, we get the water fraction in wt. Subsequently, we retrieve the composition of "H2O". This part is slightly more complex as the information of solution models (SS_vec) are stored in a different substructure with respect to pure phases (PP_vec). In order to find the right id for "H2O" in the the PP_vecsubstructure we do id_h2o - out[i].n_SS where out[i].n_SS is the total number of solution models in the stable assemblage.
Note
The previous code snipped has to be placed after calling
single_point_minimization()Mind that for the igneous database, there is a fluid model "fl" instead of pure water ("H2O").
Single point trace element partitioning
The following example shows how to perform a single point equilibrium calculation and compute trace-element partitioning, using constant, linear and non-linear values.
Let's first compute a phase equilibrium using the metapelite database:
dtb = "mp"
data = Initialize_MAGEMin(dtb, verbose=false, solver=0);
P,T = 6.0, 699.0
Xoxides = ["SiO2"; "TiO2"; "Al2O3"; "FeO"; "MnO"; "MgO"; "CaO"; "Na2O"; "K2O"; "H2O"; "O"];
X = [58.509, 1.022, 14.858, 4.371, 0.141, 4.561, 5.912, 3.296, 2.399, 10.0, 0.2];
sys_in = "wt"
out = single_point_minimization(P, T, data, X=X, Xoxides=Xoxides, sys_in=sys_in, name_solvus=true)
Finalize_MAGEMin(data)Note
The option name_solvus = true is important here, as it allows for to properly name minerals based on their composition, e.g., fsp -> pl or afs
Then we define the trace elements we want to model:
el = ["Li","Zr"]The phases for which we have partitioning coefficients (KDs):
ph = ["q","afs","pl","bi","opx","cd","mu","amp","fl","cpx","g","zrc"]Partitioning coefficient Matrix
Subsequently, we need to define the partitioning coefficients. For instance let's define some arbitrary formulations for the KD's:
KDs = ["0.17" "0.01";"0.14 * T_C/1000.0 + [:afs].compVariables[1]" "0.01";"0.33 + 0 01*P_kbar" "0.01";"1.67 * P_kbar / 10.0 + T_C/1000.0" "0.01";"0.2" "0.01";"125" "0.01";"0.82" "0.01";"0.2" "0.01";"0.65" "0.01";"0.26" "0.01";"0.01" "0.01";"0.01" "0.0"]Note
Here
KDsis a Matrix ofStringof size (n_ph, n_el).Each line of the Matrix correspond to the partitioning coefficient formulation per phase and for all elements. For instance the first line
"0.17" "0.01"indicates thatLiandZrhave KD's of"0.17"and"0.01"forq.
KDs | el_1 | el_2 | el_m |
|---|---|---|---|
| ph_1 | KD_11 | KD_12 | KD_1m |
| ph_2 | KD_21 | KD_22 | KD_2m |
| ph_n | KD_n1 | KD_n2 | KD_nm |
where n is the total number of phases and m is the total number of trace elements.
The second line is slightly more complex
"0.14 * T_C/1000.0 + [:afs].compVariables[1]" "0.01". The first entry"0.14 * T_C/1000.0 + [:afs].compVariables[1]"is a non-linear formulation ofLiKD forafsthat depends on temperatureT_Cand the compositional variable 1 of biotite. See example E.2 for details about the output structure.All variables from
MAGEMin_Coutput structure can be accessed and used to define formulation for non-linear KDs.Note that informations about specific minerals are accessed using for instance
[:afs].
We now need to define the starting concentration of the trace elements (ppm or ug/g):
C0 = [100.0,400.0]Then create the KDs database:
KDs_database = create_custom_KDs_database(el, ph, KDs)And we can finally compute trace element partitioning as:
out_TE = TE_prediction(out, C0, KDs_database, dtb; ZrSat_model = "CB")which yields:
out_TE_struct(["Li", "Zr"], [100.0, 400.0], [154.51891525771387, 3107.727391290317], [92.57069316319014, 31.017280420226772], [30.903783051542774 31.07727391290317; 262.8366748533713 31.07727391290317; … ; 26.26821559381136 31.07727391290317; 1.5451891525771388 0.0], ["opx", "bi", "pl", "q", "zrc"], [0.10855984484830646, 0.20550703811276913, 0.5068659994696709, 0.1771366556875411, 0.0019304618817123876], 0.1199276845342525, 3107.727391290317, 47.86020212957779, 0.1522004049315548, [0.5324820362432465, 0.14855391402192616, 0.05910962038616418, 0.04560199231753971, 0.043702325897822, 0.02398578811001486, 0.03295421325994538, 0.010218205689218504, 0.001999648862860764, 0.0014098120682238678, 0.0999824431430382])Trace element output structure
The out_TEstructure contains:
out_TE.
C0 Cliq Cliq_Zr
Cmin Csol Sat_zr_liq
bulk_cor_wt elements liq_wt_norm
ph_TE ph_wt_norm zrc_wtNote
C0-> the starting concentrations of the trace elementCliq-> the trace element concentrations in the liquidCliq_Zr-> the zirconium concentration of meltCmin-> the trace element concentrations in the phasesCsol-> the trace element concentrations of the solid part,Sat_zr_liq-> the melt saturation value of zirconiumbulk_cor_wt-> the corrected bulk rock composition from crystallized zirconelements-> names of thr trace elementsliq_wt_norm-> the normalized wt fraction of meltph_TE-> names of phase carrying the trace elementsph_wt_norm-> the normalized fraction of the phaseszrc_wt-> the computed wt% of crystalllized zircon
Note
Available zirconium saturation models are:
"none"-> deactivate the call to zirconium saturation model"WH"-> Watson & Harrison (1983)"B"-> Boehnke et al. (2013)"CB"-> Crisp and Berry (2022)
Trace element partitioning
First install and use ProgressMeter and Plots packages:
] add ProgressMeter, Plots
using ProgressMeter, Plots, MAGEMin_CSuch as previous example let's initialize MAGEMin using the metapelite dataset (White et al., 2014):
dtb = "mp"
data = Initialize_MAGEMin(dtb, verbose=false, solver=0);
Xoxides = ["SiO2"; "TiO2"; "Al2O3"; "FeO"; "MnO"; "MgO"; "CaO"; "Na2O"; "K2O"; "H2O"; "O"];
X = [58.509, 1.022, 14.858, 4.371, 0.141, 4.561, 5.912, 3.296, 2.399, 10.0, 0.2];
sys_in = "wt"To compute a fractional crystallization path, we can for instance choose to fix pressure:
P = 6.0 #kbarThen we need to define range of temperature for the calculation and a temperature step. This can be done as follow:
T0 = [1200.0:-10.0:600.0;]Note
- Here the
;before the]converts theVector{StepRange...}into aVector{Float64}
We can retrieve the number of steps as:
nsteps = length(T0)We can then pre-allocate the output structures to store both the results of the phase equilibrium predictions and trace element partitioning:
Out_XY = Vector{out_struct}(undef,nsteps)
Out_TE_XY = Vector{out_TE_struct}(undef,nsteps)Trace element definition
Let's now define the trace element starting conditions (as in previous example):
ZrSat_model = "CB"
el = ["Li","Zr"]
ph = ["q","afs","pl","bi","opx","cd","mu","amp","fl","cpx","g","zrc"]
KDs = ["0.17" "0.01";"0.14 * T_C/1000.0 + [:afs].compVariables[1]" "0.01";"0.33 + 0.01*P_kbar" "0.01";"1.67 * P_kbar / 10.0 + T_C/1000.0" "0.01";"0.2" "0.01";"125" "0.01";"0.82" "0.01";"0.2" "0.01";"0.65" "0.01";"0.26" "0.01";"0.01" "0.01";"0.01" "0.0"]
C_te = [100.0,400.0] #starting concentration of elements in ppm (ug/g)
KDs_dtb = create_custom_KDs_database(el, ph, KDs)Batch crystallization
Before computing a liquid line of descent, let us first calculate a batch crystallization.
First, copy the major and trace element bulk composition:
X0 = copy(X)
C0 = copy(C_te)Then perform the calculation:
@showprogress for i=1:nsteps
Out_XY[i] = single_point_minimization(P, T0[i], data, X=X0, Xoxides=Xoxides, sys_in=sys_in, name_solvus=true)
Out_TE_XY[i] = TE_prediction(Out_XY[i], C0, KDs_dtb, dtb; ZrSat_model = ZrSat_model)
endNote
The results of the phase prediction are stored in
Out_XYand trace element prediction inOut_TE_XY.Here the
Out_XY[i]is passed toTE_prediction(Out_XY[i], ...)as an argument together withC0,KDs_dtbandZrSat_model.
Available zirconium saturation models are:
"none"-> deactivate the call to zirconium saturation model"WH"-> Watson & Harrison (1983)"B"-> Boehnke et al. (2013)"CB"-> Crisp and Berry (2022)
Plot Li concentration
Let's now plot the evolution of Li concentration in the melt and save the figure.
Li_id = findfirst(isequal("Li"), el)
Li_melt = [Out_TE_XY[i].Cliq[Li_id] for i in 1:nsteps if !isnothing(Out_TE_XY[i].Cliq)]
T = [T0[i] for i in 1:nsteps if !isnothing(Out_TE_XY[i].Cliq)]Note
- Here we create
Li_meltandTarrays with the condition thatOut_TE_XY[i].Cliqis notnothingi.e., that the variable is filled. This allow to filter out values where melt is not stable.
plot( T, Li_melt,
label = "Li melt",
xlabel = "T [°C]",
ylabel = "Concentration [ug/g]",
title = "Batch crystallization of Li in a melt",
marker = :circle,
legend = :topleft,
grid = true,
xflip = true)
plot!(size=(640,400))
savefig("MAGEMin_C_batch_Li.png")which gives: 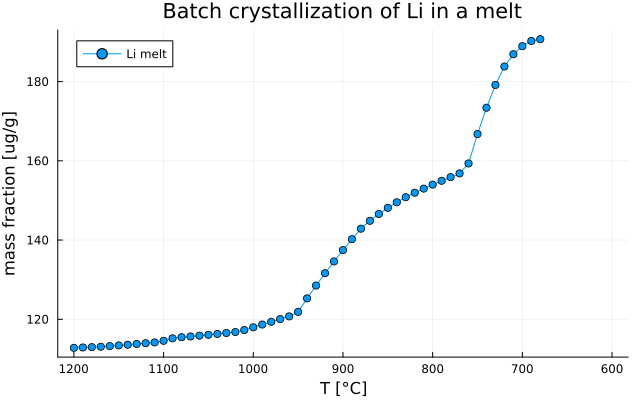
Fractional crystallization
Let's now adapt the previous code to extract cummulates during cooling.
We first need to create an additional Xoxides0 array. This is done in order to be able to update the bulk rock composition. Note that we also create a max_step variable that will record when no melt is stable anymore.
X0 = copy(X)
C0 = copy(C_te)
Xoxides0 = copy(Xoxides)
max_step = 0We then compute the fractional crystallization path:
@showprogress for i=1:nsteps
Out_XY[i] = single_point_minimization(P, T0[i], data, X=X0, Xoxides=Xoxides0, sys_in=sys_in, name_solvus=true)
Out_TE_XY[i] = TE_prediction(Out_XY[i], C0, KDs_dtb, dtb; ZrSat_model = ZrSat_model)
X0 = deepcopy(Out_XY[i].bulk_M_wt)
Xoxides0 = deepcopy(Out_XY[i].oxides)
C0 = deepcopy(Out_TE_XY[i].Cliq)
if Out_XY[i].frac_M_wt == 0.0
max_step = i
break;
end
endHere X0, Xoxides0 and C0 are updated using melt composition in wt (Out_XY[i].bulk_M_wt), the corresponding oxide list (Out_XY[i].oxides) and the trace element composition of the melt (Out_TE_XY[i].Cliq). Subsequently, the iterations are stopped if the fraction of modeled melt reach 0.0:
if Out_XY[i].frac_M_wt == 0.0
max_step = i
break;
endPlot Li concentration
We can then plot and save the results:
Li_id = findfirst(isequal("Li"), el)
Li_melt = [Out_TE_XY[i].Cliq[Li_id] for i in 1:max_step if !isnothing(Out_TE_XY[i].Cliq)]
T = [T0[i] for i in 1:max_step if !isnothing(Out_TE_XY[i].Cliq)]Note
- Here we extract
Li_meltandTup tomax_stepand notnsteps!
plot( T, Li_melt,
label = "Li melt",
xlabel = "T [°C]",
ylabel = "Concentration [ug/g]",
title = "Fractional crystallization of Li in a melt",
marker = :circle,
legend = :topleft,
grid = true,
xflip = true)
plot!(size=(640,400))
savefig("MAGEMin_C_LLD_Li.png")which gives: 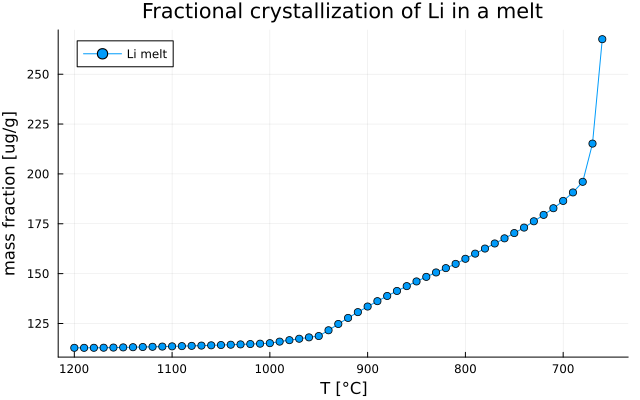
Warning
- When the bulk is first defined the order of the oxides is not necessarily as in the
MAGEMin_Coutput. When updating the bulk in a loop, for instance using the melt composition, we therefore need to retrieve the corresponding oxide ordering.
Plot fraction of zircon
Similarily we can plot the fraction of zircon crystallized from the melt
zrc_wt = [Out_TE_XY[i].zrc_wt for i in 1:max_step if !isnothing(Out_TE_XY[i].zrc_wt)]
T = [T0[i] for i in 1:max_step if !isnothing(Out_TE_XY[i].zrc_wt)]
plot( T, zrc_wt,
label = "zrc fraction",
xlabel = "T [°C]",
ylabel = "wt%",
title = "Fractional crystallization of zrc",
marker = :circle,
legend = :topleft,
grid = true,
xflip = true)
plot!(size=(640,400))
savefig("MAGEMin_C_LLD_zrc.png")which gives: 
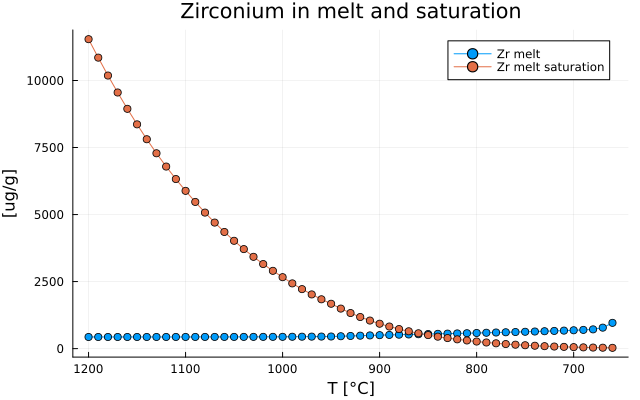
Plot Zr saturation and Zr concentration in melt
We can also plot Zr concentration and saturation of the melt as:
Zr_id = findfirst(isequal("Zr"), el)
Zr_melt = [Out_TE_XY[i].Cliq[Zr_id] for i in 1:max_step if !isnothing(Out_TE_XY[i].Cliq)]
Zr_melt_sat = [Out_TE_XY[i].Sat_zr_liq for i in 1:max_step if !isnothing(Out_TE_XY[i].Cliq)]
T = [T0[i] for i in 1:max_step if !isnothing(Out_TE_XY[i].Cliq)]
plot( T, Zr_melt,
label = "Zr melt",
xlabel = "T [°C]",
ylabel = "[ug/g]",
title = "Zirconium in melt and saturation",
marker = :circle,
legend = :topright,
grid = true,
xflip = true)
plot!( T, Zr_melt_sat,
label = "Zr melt saturation",
xlabel = "T [°C]",
ylabel = "[ug/g]",
title = "Zirconium in melt and saturation",
marker = :circle,
legend = :topright,
grid = true,
xflip = true)
plot!(size=(640,400))
savefig("MAGEMin_C_LLD_Zr.png")which gives: